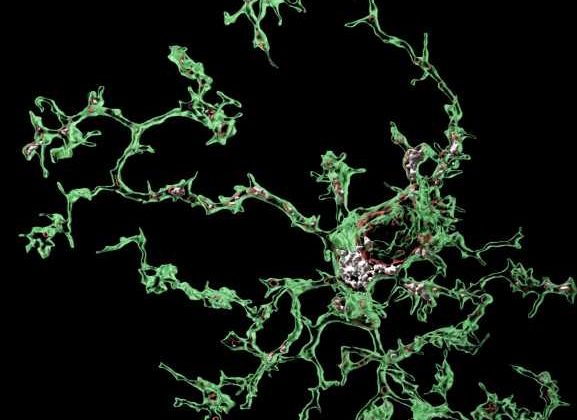

Microglia, the primary immune cells in the central nervous system, defend humans and animals from infections by “eating” pathogens, dead cells, toxic proteins and other debris in the brain. Recent neuroscience studies have consistently highlighted the role of microglia in the development of Alzheimer’s disease (AD) and other neurodegenerative diseases, suggesting that they can sometimes start “eating” or engulfing crucial connections between neurons.
Researchers at University College London (UCL) and several other institutes in the United Kingdom recently carried out a study aimed at better understanding the neural processes through which microglia can increase the risk of developing AD. Their findings, published in Nature Neuroscience, unveil the crucial role of perivascular cells, a class of cells that facilitate tissue repair, in inducing the microglial states that lead to the loss of synapses (i.e., connections between neurons) associated with AD.
“One critical function of microglia is to eat (i.e., phagocytose) or engulf various elements in the brain, including debris or pathogens, depending on what is required at the time for brain health and function,” Soyon Hong and Sebastian De Schepper, two researchers who carried out the study, told Medical Xpress. “Microglia also eat toxic aggregates that form in diseased brains and can sometimes eat critical communication junctions between neurons, called the synapses.”
While the brain is developing, microglia can “eat” or engulf improper synapses that need to be pruned away for the brain to be properly wired. Yet when the brain suffers from diseases, or when older age is causing neurodegeneration, this crucial function of microglia could go awry, leading to the engulfment of crucial neural connections.
“We were one of the first teams to show that microglia overeat synapses in a region-specific manner in AD mouse models,” Hong and De Schepper said. “This is relevant because synapse loss is one of the strongest correlates of cognitive impairment in human AD patients. Blocking microglial engulfment of synapses prevented synapse loss in AD mouse models. Importantly, this microglia-synapse engulfment pathway has been implicated in various other disease models of neurodegeneration, altogether suggesting a central role for microglia in synapse loss associated with disease. ”
In their previous work, Hong and her colleagues found evidence that microglia can overeat synapses in brains affected by Alzheimer’s disease, yet they did not yet uncover what triggers this undesirable engulfment process. The key objective of their new study was thus to investigate the regulatory mechanisms of microglia-synapse engulfment more in-depth, specifically in context of neurodegenerative diseases.
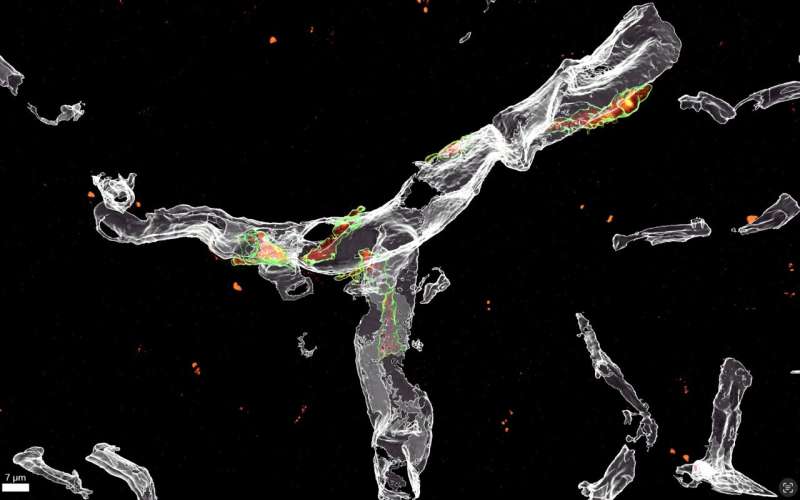
“We discovered the surprising role of perivascular cells, which reside around the blood vessels in the brain, in this process,” Hong and De Schepper said. “Using mouse models of AD, we found that these perivascular cells can influence the ability of microglia to engulf synapses. Specifically, we found that these cells secrete SPP1/Osteopontin, a glycoprotein that acts as an immune modulator, leading to activation of microglial engulfment of synapses.”
An overreaching goal of the research conducted by Hong’s lab at UCL is to pin-point the microglia subtypes contributing to the loss of synapses observed in patients with early-stage AD. Their new study was a further step in this direction, as it helped them to elucidate some of the processes involved in derailing microglia function.
“The identification and characterization of microglia subpopulations will have important therapeutic implications for the preservation of synapses in AD,” Hong and De Schepper explained. “We hypothesized that one potential subtype could be characterized by high expression levels of the proinflammatory glycoprotein SPP1 (Osteopontin). First, SPP1 marks many phagocytosing macrophage subtypes in multiple organs. In the developing brain, SPP1 marks microglia that prune excess synapses to organize neuronal networks.”
Hong, De Schepper and their colleagues also found that microglia associated with amyloid-beta plaques, the pathological hallmark of late-stage AD brains, express high levels of SPP1. This is aligned with previous studies that also reported elevated levels of SPP1 in cerebrospinal fluid and plasma of Alzheimer’s disease patients, although its cellular source had not been identified yet.
The researchers carried out a series of experiments on mice, using a combination of methods, including optical imaging and genetic techniques. To their surprise, they located SPP1 in the mice’s perivascular cells but not in microglia.
“We used a combination of optical imaging experiments including super-resolution and ultrastructural microscopy to study the different cell types that produce SPP1 in preclinical models of AD, including the AppNL-Fmouse model and acute injection model in which amyloid-beta is directly injected into the brain ventricles,” Hong and De Schepper explained. “AppNL-F mice are genetically modified to carry the App gene into their genome, mimicking typical AD features such as early synaptic pathology and amyloid plaque formation. App encodes for the amyloid precursor protein, a key component of the amyloid plaques that accumulate in the brains of people with AD.”
Hong, De Schepper and their colleagues also used genetic techniques to remove the SPP1 gene from the mice. This allowed them to determine whether the absence of SPP1 could prevent the undesired process through which microglia engulf or eat vital synapses.
Finally, the team used single-cell RNA sequencing, a transcriptomic technique used to detect active genes in individual brain cells, to determine where the SPP1 gene was located within the mice brain. Surprisingly, they located it in perivascular cells and found that it prompted them to actively instruct microglia to eat synapses.
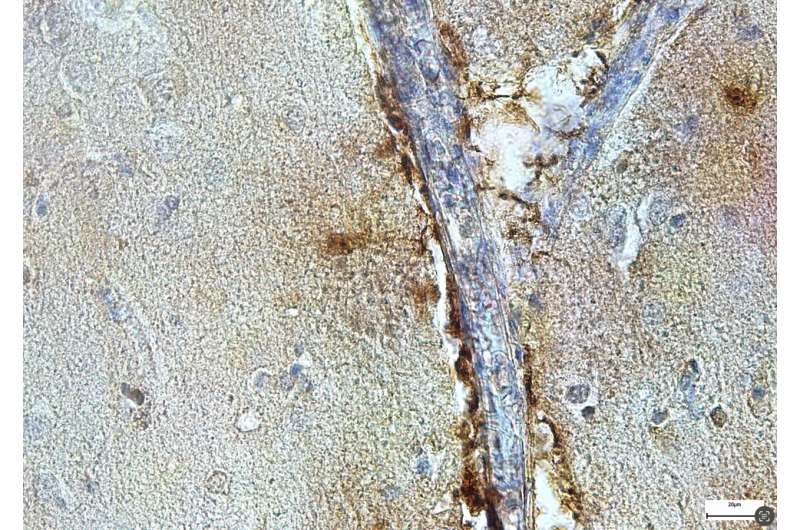
“The perivascular space is a fluid-filled structure that surrounds small blood vessels and represents the gateway between the brain and the rest of the body,” Hong and De Schepper explained. “We used transcriptomic methods to study genes in individual perivascular cells and microglia and found that SPP1 acts as perivascular cell-derived signal to instruct microglia to engulf synapses. Indeed, microglia from mice that are deficient of SPP1 (‘Spp1-Knock out mice’) were prevented from engulfing synapses, indicating that the protein is required for microglial phagocytosis to take place.”
The recent results gathered by this team of researchers confirm previous findings hinting at the central role of the vascular space in AD. Specifically, they suggest that the vascular space in the brain is a key site for the early formation of amyloid-beta protein deposits. This could in turn trigger the increased production of SPP1, which could lead perivascular cells to signal neighboring microglia, inducing the overeating of synapses associated with AD.
Interestingly, Hong, DeSchepper and their colleagues detected SPP1 in perivascular cells within the hippocampus of deceased patients with AD, meaning that their results on mice could also apply to humans. Their findings could thus soon inform the development of new therapeutic interventions for AD, which are designed to modulate microglia activity via the perivascular space.
“We are now testing different ways to therapeutically target SPP1 in the early stages of AD, aiming to prevent synapse loss and inflammation in the brain,” Hong and De Schepper added. “One way is via so-called ‘antisense-oligonucleotides’ (ASO). ASO are small pieces of genetic materials that are designed to bind to specific RNA molecules in the body. Together with the pharmaceutical company Ionis, we developed an ASO that specifically bind to SPP1 and interferes with its production, and we hope this will help to translate our findings into new therapeutic approaches for synapse preservation in AD.”
More information:
Sebastiaan De Schepper et al, Perivascular cells induce microglial phagocytic states and synaptic engulfment via SPP1 in mouse models of Alzheimer’s disease, Nature Neuroscience (2023). DOI: 10.1038/s41593-023-01257-z
Journal information:
Nature Neuroscience
Source: Read Full Article